CO TO JEST WALIDACJA?
Słownikowa definicja mówi nam, że walidacja to „badanie odpowiedniości, trafności lub dokładności czegoś”. W odniesieniu do omawianych systemów możemy przywołać definicję podaną chociażby w wytycznych do dobrych praktyk (np. wytwarzania, dystrybucji), która mówi, że walidacją jest dowiedzenie, że dany sprzęt działa właściwie i pozwala rzeczywiście osiągnąć spodziewane rezultaty.
W praktyce przed wprowadzeniem do użytku systemu skomputeryzowanego powinniśmy właśnie przez walidację udowodnić (a więc i udokumentować), że nasz system działa zgodnie z wymaganiami. Kwestia jakie są to wymagania, będzie zależała od użytkownika, gdyż może on poza wymogami stawianymi przez obowiązujące przepisy, dodać własne wymagania, których spełniania będzie oczekiwał od systemu. Sama dokumentacja powinna natomiast obejmować cele i zasady działania systemu, zakres jego działania i podejmowane środki bezpieczeństwa, a także główne jego cechy oraz sposób współpracy z innymi systemami.
SYSTEM WALIDOWANY CZY WALIDOWALNY?
Zostając dalej przy sprawach językowych chciałbym zwrócić uwagę na to, że określenia walidowany (zwalidowany) można użyć w odniesieniu do systemu, dla którego procedura walidacji została ukończona.
Wobec tego system spełniający wszystkie wymagania stawiane przez przepisy i użytkownika, przed jego zainstalowaniem powinniśmy określać jako walidowalny, czyli możliwy do zwalidowania. Warto dodać w tym miejscu, że w przypadku systemów walidowanych, na użytkowników nakładana jest szczególna troska o to aby system pozostawał zgodny z wymaganiami. Fakt, że system został w prawidłowy sposób zainstalowany i zwalidowany nie jest jednak wystarczający. W trakcie jego użytkowania niezbędne jest bowiem utrzymywanie odpowiednich procedur oraz przeprowadzanie cyklicznych rewalidacji, w zależności od krytyczności realizowanych działań.
METODOLOGIA WALIDACJI
Skoro wiemy czemu służy walidacja, pozostaje pytanie jak ją wykonać. Do wyboru mamy kilka znanych metodologii walidacji. Możemy tu wymienić modele np. wodospadowy czy kołowy. Jednak w praktyce najpowszechniej używaną i najbardziej uznaną metodologią jest GAMP (obecnie obowiązuje 5 wydanie).
Pozwala ona na praktyczne oraz oparte na analizie ryzyka przeprowadzenie walidacji. Schemat walidacji układa się na kształt litery V (od słowa validation). Poszczególne poziomy po stronie planowania łączą się z odpowiednimi dla siebie testami. Szczegółowy opis poszczególnych etapów (i stosowanych skrótów) omówię w dalszej części artykułu.
Prowadzenie testów musi być w należyty sposób dokumentowane. Przykładowo, dokumentacja dla systemu Synergy uwzględniająca wytyczne dobrej praktyki to w sumie ok. 3000 stron. Zdjęcie obok prezentuje tylko dokumentację OQ dla oprogramowania. Drugie tyle to dokumentacja dla urządzeń wchodzących w skład systemu Synergy.

Dokumentacja OQ dla oprogramowania
KATEGORIE OPROGRAMOWANIA
To jakiej kategorii oprogramowania będziemy używali będzie miało wpływ na zakres walidacji. Zgodnie z GAMP5 możemy wyróżnić 4 kategorie:
Kategoria 1 – systemy operacyjne, ogólnie dostępne oprogramowanie tj. pakiety biurowe czy arkusze kalkulacyjne
Kategoria 3 – oprogramowanie gotowe, nie konfigurowalne
Kategoria 4 – oprogramowanie konfigurowalne
Kategoria 5 – oprogramowanie dedykowane
W GAMP 4 istniała również kategoria 2 (firmware), jednak została wycofana w aktualnej wersji.
Kategoria 1 nie wymaga od użytkownika żadnych działań walidacyjnych. Zwrócić uwagę należy jednak na przykład na stosowanie formuł i makr w arkuszach kalkulacyjnych. O ile w przypadku znanych pakietów oprogramowania, nie ma obawy o działanie kodu samego programu, o tyle należy potwierdzić poprawność otrzymywanych wyników.
W przypadku systemów kategorii 3, 4 i 5 ilość testów będzie rosła wraz z numerem kategorii (dla kat. 5 najwięcej testów). Istotna jest możliwość wykonania testów fabrycznych, które będą wspólne dla określonej wersji systemu. Pozostałe muszą być dokonane w trakcie instalacji. Wobec tego w niektórych przypadkach (kategoria 3 i 4) w dalszym ciągu możemy część testów wykonać raz (np. testy OQ dla oprogramowania mogą być wykonane przez producenta i dostarczone z wypełnioną dokumentacją testów operacyjnych przeprowadzony raz, a powtarzanych dopiero przy wypuszczeniu kolejnej wersji oprogramowania). Tak więc im mamy niższą kategorię, tym mniej pracy nas czeka przy instalacji systemu.

System bezprzewodowego monitorowania warunków środowiskowych: 3 typy sond referencyjnych dające stałe wartości pomiarowe T=5°C, RH=90%; T=20°C, RH=50%; T=45°C, RH=10%.
Na zdjęciu razem z przetwornikiem, który mierzy rzeczywiste wartości temperatury i wilgotności
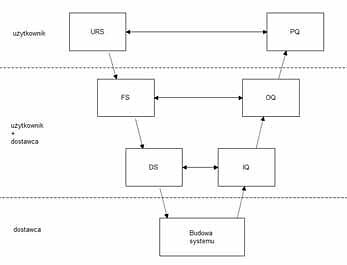
ETAPY WALIDACJI
URS (User Requirements Specification) – Specyfikacja wymagań użytkownika
Jest podstawowym dokumentem, na podstawie którego budowany jest system. Powinien zawierać szereg wymagań tj. wymagania środowiskowe, wymagania operacyjne i procesowe, wymagania prawne oraz proceduralne, a także inne wymagania techniczne. Specyfikacja wymagań użytkownika powinna być efektem przeprowadzenia rzetelnej analizy ryzyka. Jeżeli analiza taka zostanie przeprowadzona rzetelnie przed przystąpieniem do projektowania systemu, a wnioski zostaną wprowadzone do specyfikacji wymagań, to po prawidłowym ich wypełnieniu w trakcie eksploatacji, uda się uniknąć nieprzyjemnych niespodzianek. Przykładowo, wyobraźmy sobie sytuacje, w których uszkodzeniu ulegają elementy komunikacyjne lub dochodzi do zaniku napięcia zasilającego. Jeżeli weźmiemy takie sytuacje pod uwagę podczas projektowania systemu, w przypadku rzeczywistych zdarzeń nasz system powinien być na nie „odporny”. Zasadniczo wszystkie wymagania postawione w tym dokumencie powinny być spełnione, jednak w praktyce można stosować odpowiednie wagi dla niektórych kryteriów (dla przykładu: niezbędne, ważne, pożądane).
Wówczas część z kryteriów można traktować jako tzw. listę życzeń (patrząc od strony integratora często nierealną). Takim typowym wymaganiem może być niemożliwa do uzyskania dokładność urządzeń pomiarowych (przy rzetelnym podejściu do zagadnień metrologicznych).
FS (Functional Specification) – Specyfikacja funkcjonalna
Opisuje w jaki sposób (za pomocą jakich funkcji) realizowane są wymagania postawione przez URS. Możemy osiągnąć to za pomocą schematów, opisów, odesłań do dokumentacji technicznej bądź też poprzez odniesienie do konkretnych testów.
DS (Design Specification) – Specyfikacja projektowa
Definiuje budowę systemu – zarówno urządzeń jak i oprogramowania.
IQ (Instalation Qualification) – Kwalifikacja instalacyjna
Na podstawie specyfikacji projektowej, przystępując do instalacji systemu, należy ją przeprowadzić zgodnie z dokumentacją przygotowaną przez producenta. Testom powinny podlegać wszystkie komponenty poszczególnych elementów systemu. Celem tej części jest potwierdzenie prawidłowości instalacji w stosunku do zaleceń producenta i projektu.
OQ (Operation Qualification) – Kwalifikacja operacyjna
Podczas tej fazy testów sprawdzane jest prawidłowe działanie systemu pod kątem oczekiwanej funkcjonalności. Ważne jest aby podczas instalacji i testowania urządzeń, można było zarówno jednoznacznie określić poprawność działania poszczególnych elementów systemu, jak również przeprowadzić wiarygodnie symulację sytuacji awaryjnych. Można to osiągnąć różnymi sposobami, a w przypadku projektów prowadzonych przez nasz zespół, najczęściej posługujemy się tzw. sondami referencyjnym. Pozwalają one podając określone stałe wartości mierzone, na symulację pomiarów danych procesów. Umożliwia to stabilne i pewne utrzymywanie pożądanych wartości.
PQ (Performance Qualification) – Kwalifikacja działania/procesu
Jako ostatnią część kwalifikacji przeprowadza się kwalifikację procesu. Powinniśmy w niej udokumentować przede wszystkim skuteczne i powtarzalne działanie całego systemu lub urządzenia, zgodne z wymaganiami postawionymi w specyfikacji wymagań użytkownika (URS). Po zakończeniu kwalifikacji procesu, pozostaje do przeprowadzenia raport wieńczący cały proces walidacji.
WALIDACJA I CO DALEJ
Możemy zadać pytanie czy to już wszystko co powinniśmy zrobić aby nasz system był zwalidowany. Jeżeli nasze wymaganie prawne bierze się z przepisów FDA 21 CFR Part 11, w zakresie systemu skomputeryzowanego mamy nacisk położony na wiarygodność zbieranych danych oraz podpisów elektronicznych identyfikujących użytkowników.
Zwróćmy więc uwagę, że nie jest możliwe aby jakakolwiek aplikacja zapewniała wewnętrznie pełną zgodność z wymaganiami FDA 21 CFR Part11, jeśli ta aplikacja nie kontroluje całkowicie komputera PC. Ponieważ taka kontrola normalnie nie jest dozwolona w systemach korporacyjnych, reżim bezpieczeństwa powinien zostać zaplanowany i wdrożony przez dział IT danej korporacji. Pod uwagę wzięte powinny być m.in. bezpieczeństwo baz danych (w tym odpowiednio prowadzone kopie zapasowe) oraz kontrola dostępu.
W trakcie użytkowania systemu, musimy ponadto cyklicznie wzorcować przyrządy pomiarowe tworzące nasz system, a także wymieniać jego komponenty nie tylko na skutek awarii ale również normalnego zużycia. Wobec tego oczywistym jest to, że po przeprowadzeniu walidacji jedynie podczas instalacji, nie jest wystarczające aby dany system uważać za zwalidowany do końca jego użytkowania. I tutaj ponownie z pomocą przychodzi nam GAMP, który opisuje cały cykl życia systemu: od pomysłu, poprzez budowę, wdrożenie i użytkowanie, aż do wycofania z użytku. Warto zatem zaznaczyć, że podczas użytkowania, będziemy musieli cyklicznie poddawać system rewalidacji, aby mieć pewność, że dalej spełnia swoją funkcję.
Mam nadzieję, że chociaż w podstawowym zakresie udało się przedstawić temat walidacji. Mając świadomość, że w tak krótkim artykule nie jest możliwe zawarcie wszystkich niezbędnych informacji, w przypadku pytań zapraszam do kontaktu.
autor:
Paweł Szuba
pszuba@introl.pl